A Workflow for Metabarcoding of Fungi with Oxford Nanopore Sequencing
Fungi form the third-largest kingdom of life and is estimated to contain between two and three million species (Niskanen et al., 2023). To date, approximately 161,000 species have been formally described (Index Fungorum Partnership, 2024). Fungi have diverse living forms, are widely distributed and take on numerous ecological roles in terrestrial and aquatic habitats (Peay et al., 2016; Phukhamsakda et al., 2022). Saprotrophic fungi break down organic matter and are involved in nutrient cycling, while others form mutually beneficial interactions with other species such as the formation of mycorrhiza between plants and fungi (Frey, 2019). Other fungi form pathogenic interactions causing considerable damage to human health and agriculture (Fisher et al., 2020).
Metabarcoding is a technique for identifying the organisms that are present in an environment by sampling and analysing genomic content after amplification of DNA barcodes (Taberlet et al., 2018). Applications of metabarcoding include: the profiling of communities, analysis of diversity and inferring ecological patterns (Kisand et al., 2012; Corrales et al., 2021; Taberlet et al., 2018).
Fungi can be identified using morphological features of sporing (or asexual) structures or by isolating them in culture from the environment of interest (Tedersoo and Nilsson, 2016). The presence of reproductive structures is often brief and a vast number of fungal lineages (such as ectomycorrhizal fungi) are difficult to isolate in culture. DNA barcoding provides a solution to this problem by detecting the DNA of targeted organisms without direct observation (Hajibabaei et al., 2011).
Advances in high-throughput sequencing (HTS) has provided the capacity to identify many species from bulk environmental samples such as soil (Tedersoo et al., 2022). This procedure (metabarcoding) works by extracting the genomic DNA of all organisms within a sample, followed by the amplification of a barcode region of interest using the polymerase chain reaction (PCR) (Lindahl et al., 2013). The amplified DNA is sequenced by a HTS platform such as Illumina, PacBio or Oxford Nanopore Technologies. Using computational techniques, sequences can be analysed and compared to those in reference databases to identify taxa. The accuracy of these taxonomic classifications depends heavily on the quality of the reference data used (Tedersoo et al., 2022).
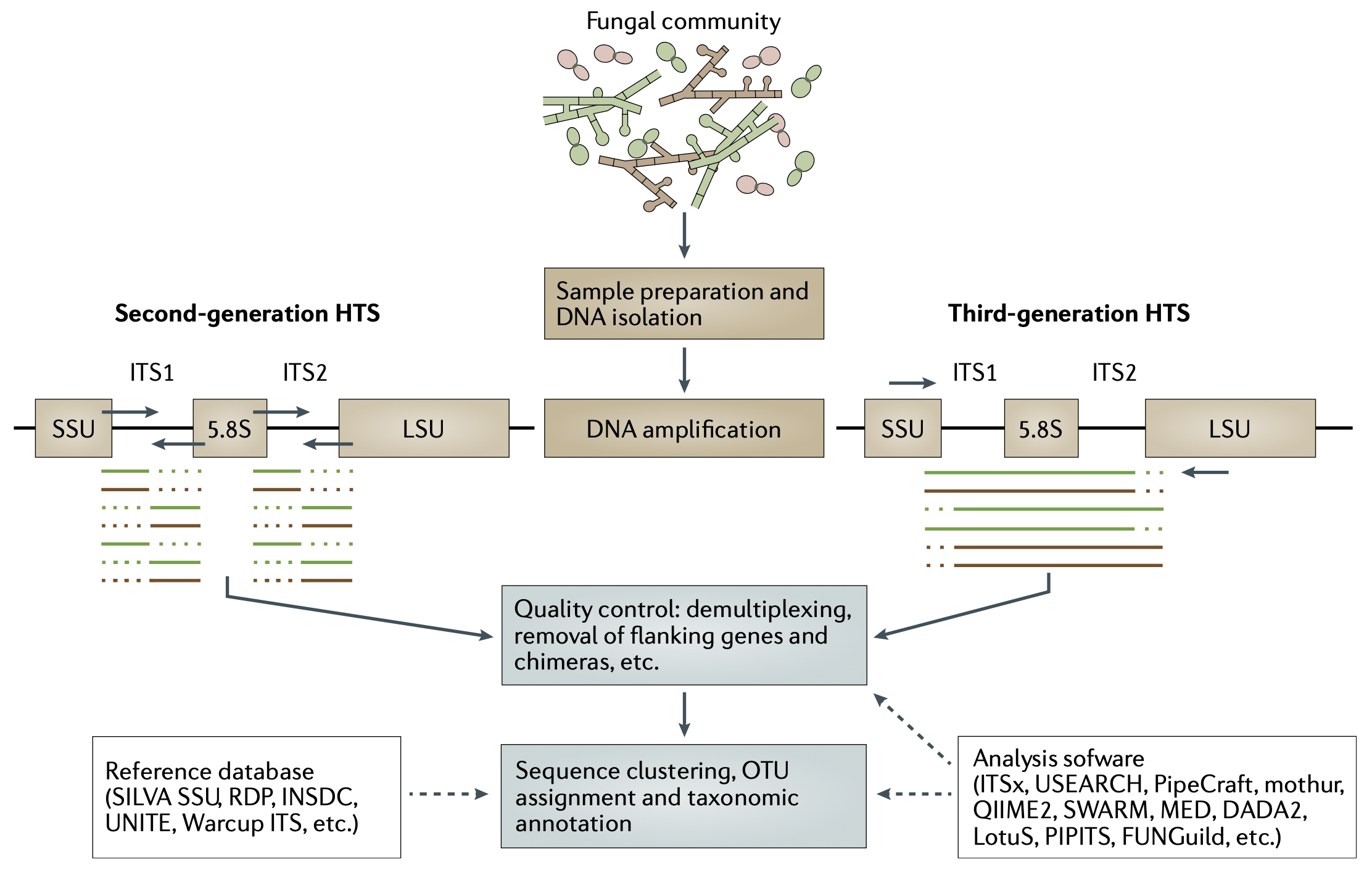
The barcode region most commonly used for metabarcoding of fungi is the internal transcribed spacer (ITS) region which is found between the 18S and 28S subunits of the nuclear ribosome, also known as the small subunit (SSU) and large subunit (LSU) respectively (Tedersoo et al., 2022). The ITS region has become the universal marker for barcoding fungi for multiple reasons. PCR amplification of the ITS marker has a high success rate due to highly conserved regions which flank the marker. This allows PCR primers can be designed to cover a high range of Fungi (Schoch et al., 2012). See Figure 1 for a general overview of fungal metabarcoding. As ITS regions are non-coding, they are highly variable and provide high inter- and intraspecies resolution (Schoch et al., 2012).
HTS technologies have been adopted for metabarcoding due to large sequencing capacity and the ability to characterize DNA sequences from mixed samples as opposed to traditional methods such as Sanger sequencing (Nilsson et al., 2019). Short-read HTS methods such as Illumina MiSeq are commonly used for metabarcoding studies and allow for the sequencing of up to 2x300 (forward and reverse direction) basepairs (bp) per read (Tedersoo et al., 2022; Illumina Inc., 2023). In fungal metabarcoding, these read length limitations makes Illumina best suited to analyse the smaller ITS1 or ITS2 subregions separately with a very high sequencing depth (Nilsson et al., 2019). Illumina MiSeq, NextSeq and HiSeq produce a very high numbers of reads per run (up to 25 million, 1.2 billion and 6 billion respectively) that result in high quality denoised sequences (Illumina Inc., 2023, 2016).
Sequencing platforms such as Oxford Nanopore Technologies (ONT) and PacBio provide much longer read lengths compared to Illumina HTS methods. ONT sequencing is comparatively cheap and can be performed in a basic molecular lab or in the field (Mafune et al., 2020; Baloğlu et al., 2021). Until recently, the high error rate made ONT sequencing unsuitable for metabarcoding studies (Zhang et al., 2023). The prospect of long-read sequencing for fungal metabarcoding is that the longer reads will be able to cover multiple marker genes, including the full ribosomal operon, and can provide more reliable phylogenetic placement and discovery of new fungal lineages (Tedersoo et al., 2018).
In the existing research, studies have explored the use of ONT sequencing for metabarcoding of soil microbial community structure (Wiryawan et al., 2022), rapid pathogen detection (Hong et al., 2020; Ohta et al., 2023), and testing the use of ONT for metabarcoding of mock fungal communities (Baloğlu et al., 2021; Mafune et al., 2020; Langsiri et al., 2023). In this study, we integrated common approaches to fungal metabarcoding into a bioinformatic workflow with the focus on analysing ONT data. We validated our metabarcoding approach with mock fungal communities and tested the workflow on real environmental soil samples.