2 Literature Review
1 Fungal barcode regions
Molecular barcoding is a technique for identifying organisms using a unique DNA region (or barcode) that is common to the group of targeted organisms (Taberlet et al., 2018). The variability of a DNA sequence among species provides the ability to discriminate between taxa (Hebert et al., 2003). For an ideal molecular barcode, the interspecific variation is much higher than the intraspecific variation (Schoch et al., 2012). The difference between these variations is called the barcode gap (Puillandre et al., 2012). A barcode is optimal when the sequence is unique to one species and there is little variability of the barcode within that same species (Schoch et al., 2012).
In fungi, barcode regions most often used for species identification are those that encode the nuclear ribosomal RNA (rRNA) and more rarely from other regions that encode proteins (Schoch et al., 2012). The nuclear rRNA region contains the 18S small subunit (SSU), 5.8S and 28S large subunit (LSU) rRNA genes. These genes are transcribed by RNA polymerase I and two spacer regions (ITS1 and ITS2) found between the genes are removed through post-transcriptional modifications. The region covering the two spacers is referred to as the internal transcribed spacer (ITS) region and typically spans 500–700 bases but is highly variable among fungal groups (Figure 1, Nilsson, Anslan, et al. (2019)).
.png)
Due to read length limitations of short-read sequencing platforms, generally only one of the ITS1 or ITS2 subregions (250-400 bases) are targeted for sequencing (Nilsson, Anslan, et al., 2019). The limitations of using the ITS1 or ITS2 subregions in isolation is that they have lower taxonomic resolution and universal PCR primer sites are less convenient than the full ITS region (Tedersoo et al., 2022, 2015).
The ITS region has limitations on its coverage of taxa and is unsuitable for some groups of fungi. For example some species in the Microsporidia group may lack the ITS region, and other orchid root symbionts (Tulasnellaceae) have mutations in primer sites (Tedersoo and Nilsson, 2016; Tedersoo et al., 2022). The arbuscular mycorrhizal Glomeromycota have highly variable copies of the ITS region, with up to 20% divergence in a single multinucleate spore (Schoch et al., 2012). For these reasons, metabarcoding studies targeting arbuscular mycorrhizal fungi commonly use SSU or LSU instead of ITS (Delavaux et al., 2022; Öpik et al., 2010). Other fungal genera such as Trichoderma and Fusarium lack variability, making the ITS region unsuitable to discriminate between species (Tedersoo et al., 2022).
Copy numbers of the ITS region can vary between species and non-identical copies can co-exist within the same individual (Bradshaw et al., 2023). This can lead to amplification bias, inflating diversity in organisms with higher copy numbers (Nilsson, Anslan, et al., 2019). Despite these limitations, ITS has a much higher PCR amplification success rate compared to protein-coding genes such as RPB1, RPB2 and MCM7 and is therefore better suited for metabarcoding (Schoch et al., 2012).
Other rDNA genes used as fungal barcodes are the 18S SSU and 28S LSU. Since they encode RNA involved in ribosome formation, these regions are highly conserved and there is often insufficient variation in the SSU and LSU regions to discriminate between many basidiomycete and ascomycete species (Schoch et al., 2012). Nevertheless, the SSU and LSU markers can be useful at higher taxonomic levels to resolve the evolutionary history of genera, families, order and class (Nilsson, Anslan, et al., 2019).
2 Sequencing methods
Molecular identification strategies have had a significant impact on fungal ecology research and have allowed investigators to identify organisms by sequencing their DNA. Early molecular studies in the 1990s indicated that there were many unknown fungal species that could not be detected from sporing bodies, isolation in culture or other observable structures (Gardes and Bruns, 1996; Nilsson, Anslan, et al., 2019; Tedersoo and Nilsson, 2016).
The high throughput sequencing (HTS) technologies 454 pyrosequencing and IonTorrent emerged in the 2000s that allowed for automated sequencing of mixed samples, enabling the identification of tens to hundreds of taxa from multiple samples (Jumpponen and Jones, 2009). The rapid adoption of 454 pyrosequencing led to an influx of sequencing data and a boom of the fungal ecology field (Hibbett et al., 2009). The 454 pyrosequencing platform was primarily adopted for fungal ecology studies between 2008 and 2014 and was capable of producing 1.2 million reads with a length of up to 1000 bases (Nilsson, Anslan, et al., 2019). Drawbacks to the pyrosequencing technique were the high cost and difficulties in sequencing homopolymer-rich regions (Tedersoo and Nilsson, 2016). As of 2016, the production of 454 sequencers was discontinued (BioIT, 2013).
Ion Torrent technology produced similar quality results to 454 pyrosequencing at lower cost but was limited to shorter read lengths (400–600 bp) and complex library preparation protocols (Tedersoo and Nilsson, 2016). Due to these drawbacks, Ion Torrent was never broadly used in fungal ecology (Nilsson, Anslan, et al., 2019).
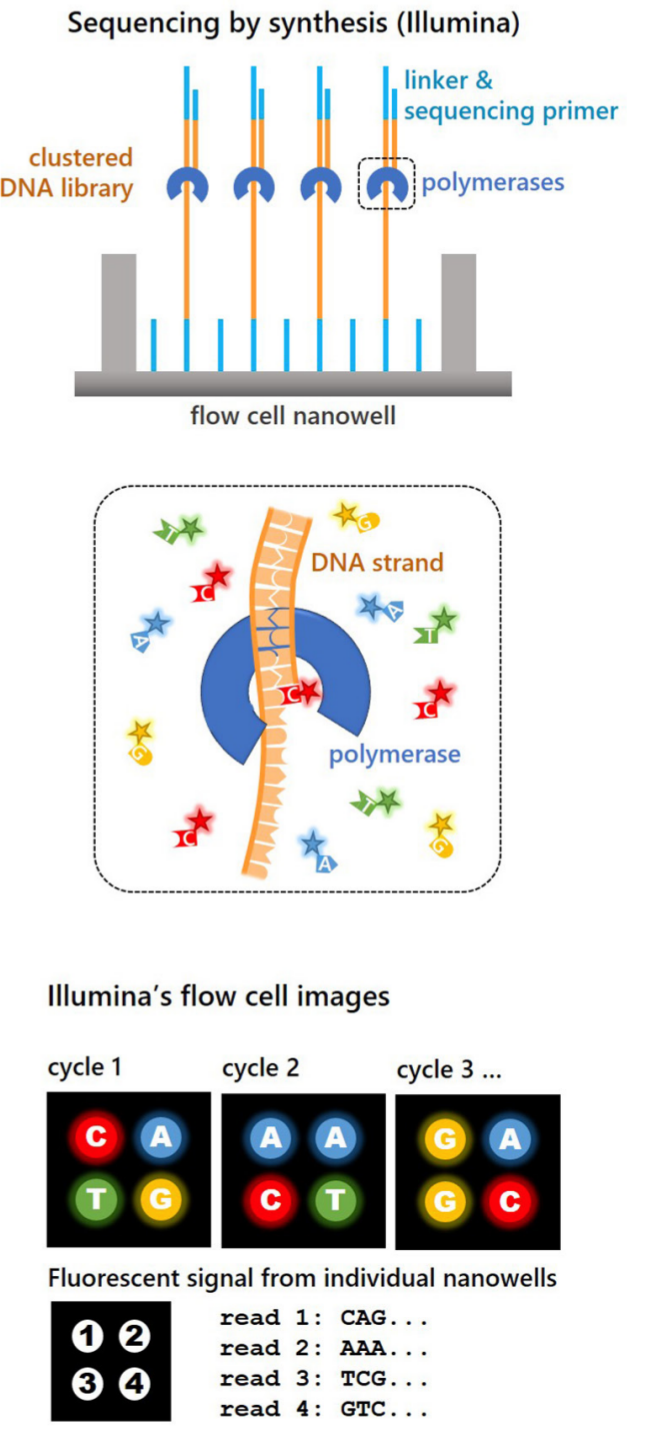
The next advance in sequencing which had wide adoption was Illumina MiSeq technology (Tedersoo and Nilsson, 2016). The Illumina MiSeq, (and more recently) NextSeq and NovaSeq platforms offer short paired-end reads of up to 300 bp in length. These platforms provide very high throughput and high quality sequences. MiSeq, NextSeq and NovaSeq can sequence up to 20, 360 and 6000 gigabase (GB) of read data respectively (Nilsson, Anslan, et al., 2019; Illumina Inc., 2023). Of the sequencing systems, Illumina can provide one of the highest read qualities with typical accuracy of 99.9% (Illumina Inc.). The use of paired end reads (on forward and reverse strands) can enable the coverage of a longer ~550 base region (Nilsson, Anslan, et al., 2019). For these reasons, the Illumina sequencing platforms are currently the standard for use in metabarcoding and metagenomics (Nilsson, Anslan, et al., 2019).
Recent advances in sequencing technology have provided much longer reads and are capable of sequencing a single DNA molecule, as opposed to clusters of DNA molecules as in Illumina sequencing (Figure 2) (Eid et al., 2009; Aigrain, 2021). Until recently, the low read quality of these techniques has been a limitation for metabarcoding studies (Nilsson, Anslan, et al., 2019).
The PacBio HiFi sequencing platform can provide read lengths of 10–25 KB and improves error rates through circular sequencing which allows the same molecule to be sequenced repetitively (PacBio, 2023; Tedersoo and Nilsson, 2016). PacBio HiFi reads can reach a base call accuracy comparable to Illumina of 99.9% but the relatively high cost is a major drawback for its adoption (Aigrain, 2021; Murigneux et al., 2020).
Oxford Nanopore Technology’s (ONT) sequencing platform has gained much attention in recent years and has been observed to produce read lengths of up to 2.4MB (Payne et al., 2019). With the ONT platform, sequencing can be performed at a relatively low cost and is much more accessible than other platforms since it can be run from a laptop, even in the field (Mafune et al., 2020). Until recently, a large drawback of the ONT system was the high raw error rate which had been reported to range between 1% and up to 15% (Baloğlu et al., 2021; Rang et al., 2018). Techniques to improve accuracy are advancing with new sequencing chemistry and basecalling algorithms. ONT is now claiming a raw read accuracy rate of 99.75% with the latest chemistry (Oxford Nanopore Technologies, 2024a) which is comparable to Illumina MiSeq.
ONT sequencing works by threading a single strand of DNA (or RNA) through a pore with an internal diameter on the order of ~1.4nm to 2.4nm (Wang et al., 2021). The pore is situated on a charged membrane and allows ions to flow across the membrane. The sequencing of base pairs is made possible by analysing the change of electric current across the membrane as the DNA (or RNA) molecule is ratcheted through the pore (Figure 3).
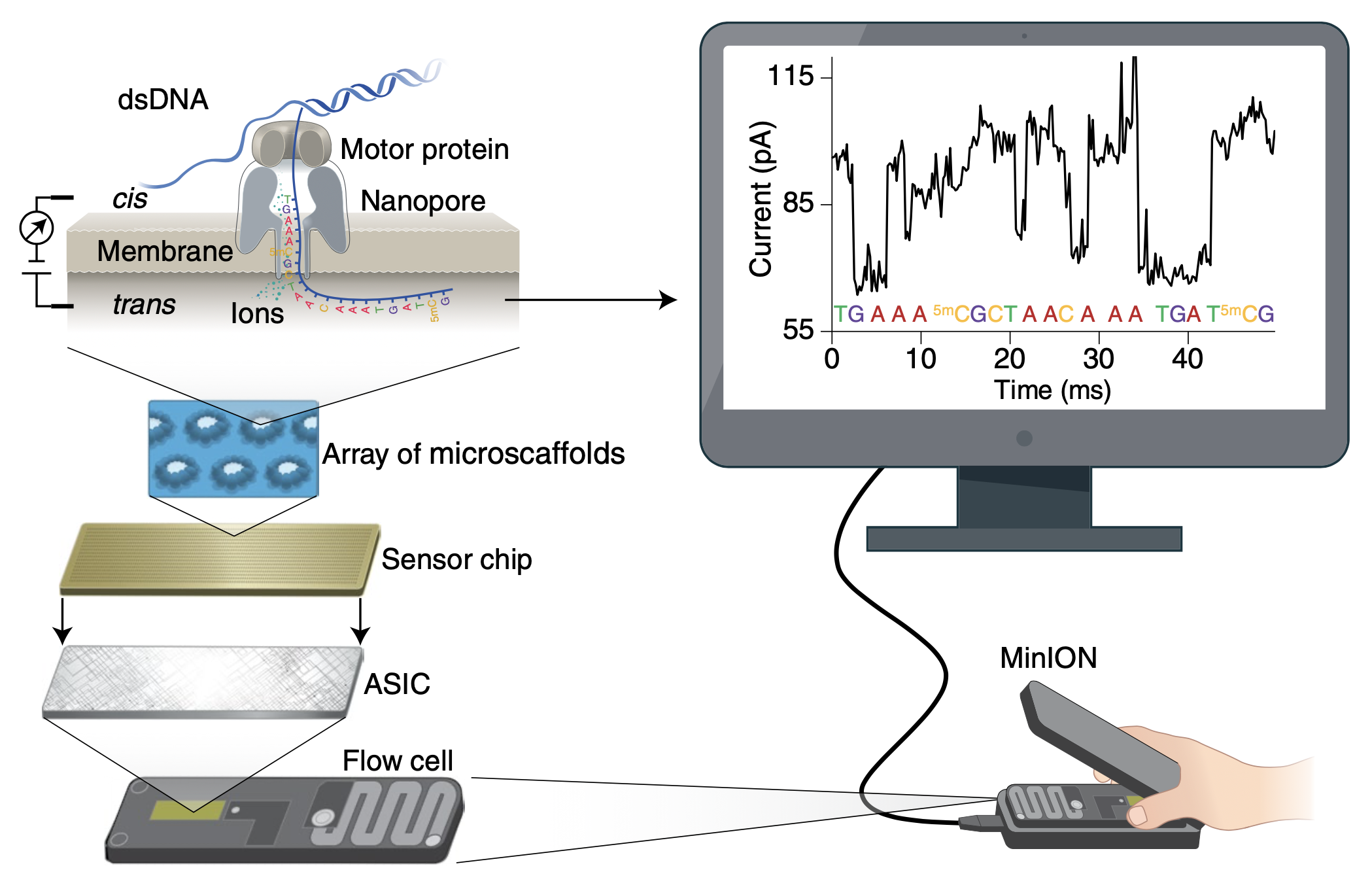
ONT’s first product was the smartphone sized MinION, which was released in 2014 (Oxford Nanopore Technologies, 2023). Improved versions of their nanopore and motor proteins have been iteratively released starting with version R6 (in 2014) and their latest being R10.4.1 (Wang et al., 2021; Oxford Nanopore Technologies, 2024b). It is worth noting that none of the specifications for the proteins used in these systems have been fully disclosed by ONT (Wang et al., 2021).
Basecalling is the process where raw electrical signals from the device are analysed to determine the sequence of nucleotides. The electrical signal is influenced by the nucleotides that occupy the pore at a given time. Historically, basecallers used hidden markov models (HMMs) to infer a DNA sequence from an electrical signal but modern basecallers now adopt neural networks and accuracy has improved significantly with each iteration (Wick et al., 2019; Rang et al., 2018). ONT’s latest basecaller is dorado which is: open source, optimised for graphics processing units (GPUs) and replaces ONT’s previous (now unsupported) basecaller guppy (Nanoporetech/dorado, 2024).
3 Bioinformatic analysis
Here we will outline the key computational steps common to many metabarcoding analyses as outlined in Figure 1.
3.1 Quality filtering
Quality filtering is an initial step in a metabarcoding pipeline that processes the raw output of the sequencing instrument for further analysis and involves: demultiplexing, trimming of primer and index sequences, and removal of low quality and off target reads (Tedersoo et al., 2022).
Multiplexing is a method for reducing costs by sequencing multiple DNA samples simultaneously, where each sequence contains a tag (or barcode) that is unique to the sample it belongs to (Wick et al., 2018). Demultiplexing is the process of splitting reads by these tags to determine which sample the read came from (Wick et al., 2018). The ONT basecallers dorado and guppy have built-in support for demultiplexing, while minibar (California Academy of Sciences, 2024) is suitable for custom barcoding schemes. Historically, custom tools such as Porechop (Wick) were developed to trim ONT sequencing adapters from reads, but currently this functionality is supported by dorado.
For ITS metabarcoding, it is useful to remove the genes flanking the ITS1 and ITS2 subregions (such as 18S, 5.8S and 22S) as they are more conserved and do not enhance species level resolution (Nilsson, Anslan, et al., 2019; Tedersoo et al., 2022). The tools ITSx (Bengtsson-Palme et al., 2013) and ITSxpress (Rivers et al., 2018) are capable of extracting ITS1, ITS2 and full length ITS regions using hidden markov models. ITSxpress expands on the functionality of ITSx, targeting its use for amplicon sequence variant (ASV) techniques over OTU clustering (Rivers et al., 2018).
To ensure the sequence data is usable for downstream analysis, quality filtering tools remove entire reads that do not meet a quality threshold or trim low quality segments from reads (Taberlet et al., 2018; Hakimzadeh et al., 2023). Some considerations are needed when performing quality filtering for ONT data. Tools such as NanoFilt (De Coster et al., 2018) and Chopper (De Coster and Rademakers, 2023) accept or reject an entire read based on an average quality score threshold. For ONT reads with variable quality, stretches of the read with high sequence quality may still be salvaged using Prowler (Lee et al., 2021) or Filtlong (Wick).
3.2 Clustering
In general, clustering is an unsupervised technique for grouping items together based on their similarity to each other. In the context of metabarcoding, clustering is the process of aggregating DNA sequences into groups based on a predefined similarity metric.
In metabarcoding, a commonly used similarity metric is sequence identity and is defined as the proportion of nucleotides that match exactly between two sequences. Sequences are considered to be in the same cluster when the pairwise sequence identity is above a predefined threshold. The resulting clusters are called Operational Taxonomic Units (OTUs) (Cline et al., 2017; Blaxter et al., 2005). For fungal metabarcoding, sequence identity thresholds of OTUs usually range from 95% to 100%, most often delineating species at 97% (Tedersoo et al., 2022; Joos et al., 2020). Three types of clustering frequently seen in metabarcoding are: closed reference, de novo and open reference clustering (Tedersoo et al., 2022; Cline et al., 2017).
Closed reference clustering is a method where OTUs are constructed based on similarity to sequences in a reference database (Cline et al., 2017). As a drawback, the biological variation that is not represented in the reference database is lost when clustering with this method (Callahan et al., 2017). Closed reference clustering allows OTUs to be tracked across studies provided the same reference database is used (Cline et al., 2017; Callahan et al., 2017).
De novo clustering is a commonly used method for defining fungal OTUs where sequences within an experimental dataset are clustered without relying on a reference database (Cline et al., 2017). The OTUs generated by de novo clustering are dependent on the dataset in which they are defined. Therefore, de novo OTUs clustered from different datasets cannot be reliably compared (Callahan et al., 2017). Compared with closed reference clustering, de novo clustering produces less stable OTUs but can detect biological variation that is not present in a reference database (He et al., 2015).
Open reference clustering combines the benefits of closed reference and de novo methods. Sequences are first clustered based on similarity to those in a reference database followed by de novo clustering of the unassigned sequences (Cline et al., 2017).
The amplicon sequence variant (ASV, also called the exact sequence variant) approach differs significantly from traditional OTU clustering by aiming to correct errors in reads then grouping identical reads together. ASV methods discriminate biological sequences from errors on the expectation that errors are rare (Callahan et al., 2017). ASV methods can detect small biological sequence variants, such as infraspecific variation (i.e. between populations within a species), while disregarding errors introduced through sequencing or library preparation, therefore increasing the taxonomic resolution of the results (Joos et al., 2020; Glassman and Martiny, 2018). Compared to OTU clustering where identity thresholds are defined artificially, ASVs have the advantage to be more precise and reproducible (Callahan et al., 2017). ASVs are largely used in metabarcoding studies targeting conserved regions such as the bacterial 16S. However, in fungal metabarcoding, ASVs are expected to perform poorly for groups that have multiple ITS copies and variants within individuals (Tedersoo et al., 2022). Therefore, ASV approaches tend to overestimate richness of common fungal species (due to haplotype variation) and underestimate richness of rare species (by removing rare variants) (Tedersoo et al., 2022; Joos et al., 2020). Nevertheless, when analysing diversity patterns based on dominant members of fungal communities, results are usually similar between ASV and OTU based approaches (Glassman and Martiny, 2018).
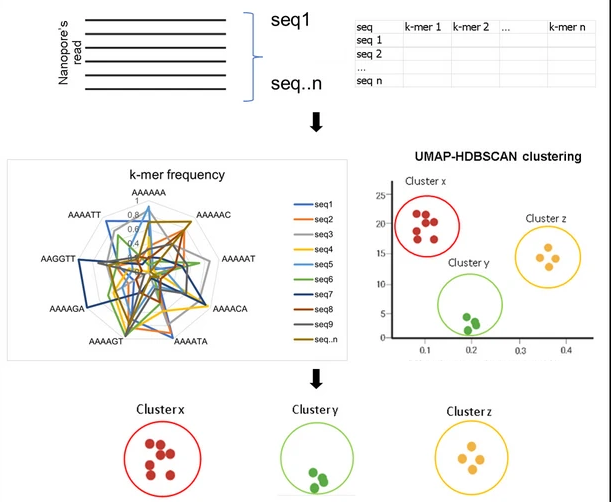
A completely different approach to clustering has been implemented in the NanoCLUST pipeline (Rodríguez-Pérez et al., 2021) which groups sequences by k-mer signature similarity. First, each sequence is transformed into a k-mer frequency vector and stored in a tabular format (one column per k-mer). A dimension reduction step converts the multidimensional tabular structure into two dimensions using Uniform Manifold Approximation and Projection (UMAP) (McInnes et al., 2020). The similarity of sequences is represented as distances between points in two-dimensional space. The Hierarchical Density-Based Spatial Clustering of Applications with Noise (HDBSCAN) algorithm is then used to cluster these points (Figure 4, McInnes et al. (2017)). HDBSCAN is a dynamic clustering approach that takes into consideration the density surrounding points which allows it to detect and remove noise. HDBSCAN requires setting a minimum cluster size parameter (min_cluster_size) which controls the minimum number points that are required to be considered a cluster. The parameter has the effect of filtering out small clusters or merging nearby clusters together (Leland McInnes et al., 2016). NanoCLUST has been developed for the analysis of 16S rRNA amplicons but other researchers have had success applying the same approach to fungal ITS sequences (Langsiri et al., 2023).
3.3 Taxonomic assignment
In fungal metabarcoding, taxonomic assignment is the process of classifying sequences to species or higher taxonomic ranks (Taberlet et al., 2018). If the sequences have been clustered into OTUs, a representative sequence from each OTU is used for the classification (Tedersoo et al., 2022). Sequences can also be given a taxonomic assignment directly without clustering (Klaubauf et al., 2010; Nygaard et al., 2020).
The quality of the taxonomic assignment depends on the reference database and algorithm used (Furneaux et al., 2021). Public INSDC databases such as GenBank (www.ncbi.nlm.nih.gov/genbank) are problematic for taxonomic assignment as a significant number of entries are incorrectly identified at the species level (Nilsson et al., 2006; Bidartondo, 2008). Curated databases such as UNITE (Kõljalg et al., 2005) address this problem by maintaining high quality reference records of ITS sequences and curating taxonomic designation by researchers well versed in the taxonomic groups (Abarenkov et al., 2010). Similarly, the Ribosomal Data Project (RDP) (Cole et al., 2014) provides curated fungal LSU sequences while Silva (Quast et al., 2013) provides both SSU and LSU reference sequences. A drawback of curated databases is that they have limited taxonomic coverage and do not always include the most recently published sequences (Furneaux et al., 2021).
A common method for taxonomic assignment is a Basic Local Alignment Search Tool (BLAST) search (Camacho et al., 2009), where pairwise comparisons are made between representative sequences and those in a reference database to assign the closest matching reference (Taberlet et al., 2018; Tedersoo et al., 2022). The BLAST algorithm can massively reduce search space by scanning a database of sequences for small, fixed size subsequences (knows as words). These matching sites are used as starting points to initiate more in-depth alignment. Fixed BLAST percent identity cutoffs have often been used to classify sequences at a given taxonomic rank (i.e. 98%, 90% and 85% sequence identity to assign species, genus and family (Tedersoo et al., 2014)). However, this approach is restrictive because fixed thresholds that accurately delineate taxa in one part of the fungal tree of life may not be accurate for other clades in the tree (Vu et al., 2016, 2019; Vu et al., 2022). Dnabarcoder is a tool that has been developed to address this issue by predicting similarity cutoffs for different fungal clades in a reference sequence dataset and can also classify sequences (via BLAST) using these predicted cutoff (Vu et al., 2022).
Minimap2 (Li, 2018) and Kraken2 (Wood et al., 2019) can be used as alternatives to BLAST for taxonomic classification. They both use minimisers (Roberts et al., 2004) to improve performance and handle long, potentially noisy reads.
Alternatively, there is an expanding field of machine learning (ML) techniques to classify sequences. Generally, ML techniques optimise a computer model to correctly identify sequences from a training set (where the taxonomic identity of sequences are known). The trained model is then used to classify new, unseen sequences. ML techniques will not be discussed in detail here but some notable tools for taxonomic classification are: the Naive Bayesian Classifier (Wang et al., 2007), SINTAX (Edgar, 2016) and IDTAXA (Murali et al., 2018). Recent neural network based classification methods such as MycoAI (Romeijn et al., 2024) show promise in rapid performance with comparable accuracy to other classification methods.
Phylogenetic placement algorithms such as pplacer (Matsen et al., 2010) and EPA-ng (Barbera et al., 2019) place amplicon reads onto a pre-established phylogenetic tree. They are not well suited for ITS sequences due to the high variability in length and content that produces alignments with low accuracy, but work well with more conserved regions such as 16S, SSU and LSU (Furneaux et al., 2021; Tedersoo et al., 2022).
High throughput sequencing techniques applied to metabarcoding have detected a high diversity of fungal taxa that are undescribed and entire groups (even phyla) that cannot be placed within the current fungal tree of life (Nilsson, Larsson, et al., 2019; Phukhamsakda et al., 2022). These so-called "dark taxa" do not seem to form discernible morphological structures or be cultivatable in the lab, therefore dark taxa are predominantly detected by DNA sequencing (Nilsson et al., 2023). In some cases of environmental metabarcoding, dark taxa make up the majority of taxa recovered (Retter et al., 2019). Long-read sequencing technology is being used to help phylogenetic placement for previously undescribed sequences by capturing the SSU and LSU genes in combination with the ITS region and improving taxonomic resolution (Tedersoo et al., 2020). Taxa known only from sequence data poses a dilemma for the field of mycology as the existing taxonomic system requires morphology-based classification (James et al., 2020). This trend is expected to continue as the cost of sequencing decreases and the volume of undescribed sequences continues to grow and outnumber the fully described taxa (James et al., 2020).